前言
在这一节中我们将学习基因集分析(gene set analysis)背后的统计学原理。在这一部分里,我们要记住三个重要的步骤,这三个步骤都非常重要,但它们又相互独立,分别如下所示:
- 参与统计的基因集
- 汇总每个基因集的统计学指标
- 评估不确定性的方法
准备数据与基因集数据库
|
|
加载以下包:
|
|
我们使用下面包中的数据来说明一下,这个包可以从Github中下载,如下所示:
|
|
我们构建的这个数据含有批次效应,因此我们需要使用SVA对其进行校正,如下所示:
|
|
我们需要注意一下,在这一节中,我们需要每个基因的评分和差异表达基因基因的候选列表。我们如何获取这个候选基因列表与所表示的基因丰度的单位无关。为了说明这一点,我们会假装我们事先不知道在不同组之间基因表达的关系。
我们构建一个FDR为25%的候选基因列表。这个列表中有23个基因。我们从中能了解到什么生物学信息呢?既然我们知道基因存在着相互作用,那么研究一组基因,而不是单个基因有着重要意义。通常来说,这就是基因集分析的原始想法。
网上有各种提供基因集的资源。这些资源中的基因都是经过缜密筛选的,它们都有着一些共同的特征。这些基因分组的方式是通过通路(例如KEGG)或GO来划分的。在下面的网站上就能找到许多这样的资源:
http://www.broadinstitute.org/gsea/msigdb/index.jsp
在以下的案例中,我闪将使用一个根据染色体进行分组的基因集来分析数据。第一步就是下载数据库,不过你需要在网站注册。
当你下载好文件后,就可以通过以下的命令读入数据库(文件扩展名为gmt,其实就是GSEA分析中的基因集文件),如下所示:
|
|
这个对象中含有326个基因集。每个基因集使用ENTREZ ID标明了基因,如下所示:
|
|
我们可以使用以下方式获取ENTREZ ID,如下所示:
|
|
现在,为了将其应用于我们的数据,我们必须将这些ID映射到Affymetrix ID上。现在我们编写这样一个函数,这个函数会使用Expression Set类,如下所示:
|
|
基于相关检验的方法
现在我们就可以计算差异表达基因是否在某个基因集中富集。那么我们如何计算呢?最简单的方法就是独立卡方检验,这种统计方法我们以前学过。现在我们对这些差异基因是否在Y染色体上的基因集中富集进行计算,如下所示:
|
|
因为我们比较的是男性与女性,因此我们使用Y染色体上基因集来进行富集,这样比较便于富集差异基因,更能说明这种算法的原理。但是,由于只有13个基因有显著性(在Y染色体上的只有4个),这种方法对于寻找可能在某些方面具有生物学意义的基因集来说,实际意义不大。例如,X染色体上的基因集是我们感兴趣的基因集么?这里需要注意的是,一些基因在X染色体上并不活性,因此与男性相比,女性体内这样的基因数目更多。
这里我们面临的问题是,FDR值为0.25,这是任意设置的一个截止值。为什么不设为0.05或0.01呢?我们如何设置这个截止值?
如下所示:
|
|
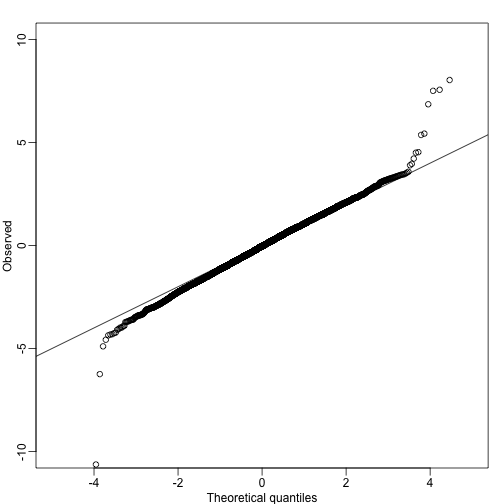
基因集相关的统计学
目前大多数进行基因集分析的方法都没有将基因分析为差异表达部分和非差异表达部分。相反,我们会使用一些类似于t检验的方法来进行计算。为了了解这种方法比基因列表的二分法更为强大,现在我们提出一个问题:一些基因是否在X染色体上并没有失活?我们不像前面提到的那样创建一个基因表,而是查看一下X染色体中第一组基因t检验中t值的分布,如下所示:
|
|
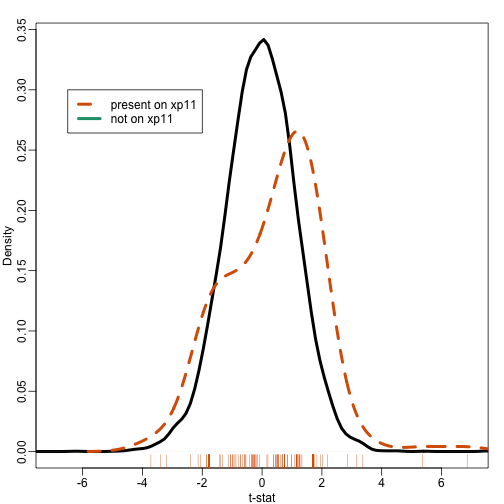
从上图我们可以看到基因集的t统计量的分布,它的分布似乎不同于所有基因的分布。那么我们如何地这个结果进行定量呢?
有关基因集分析的第一篇文献是于2001年由[Virtaneva]发表的,他在前面部分中讨论了Wilcoxon Mann-Whitney检验。那篇文献的基本思路就是统计实验组和对照组之间的差异基因在某个特定的基因集中的数目是否比在其它基因集中的数目更大。Wilcoxon比较的是基因集上基因的秩是否在中位数以上。
在这里我们计算了每个基因集的Wilcoxon检验。然后我们对Wilcoxon统计量的结果进行标准(standardize),也就是将它们转换为均值为0,标准差为1的数据。注意,中心极限理论告诉我们,如果基因集足够大,并且每个基因的效应大小彼此独立,那么这些统计量将遵循正态分布,如下所示:
|
|
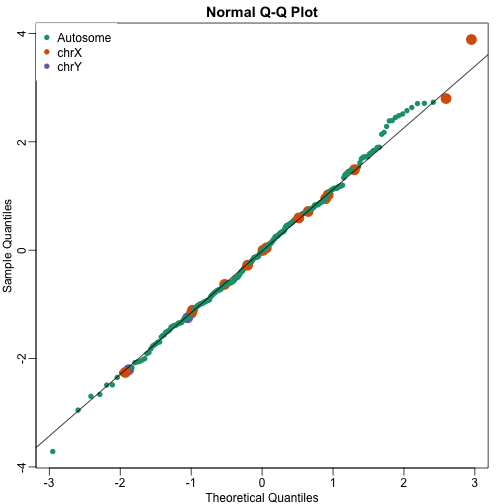
从上图我们可以看出来,排名前10位的基因并没像我们预期的那样在X染色体和Y染色体中高表达。使用Wilcoxon(稳健,robust)为基础的背后逻辑是,我们并不想看到一个或两个高表达的基因对基因集分析产生重要影响(Wilcoxon检验是功效比较低,但比较稳健,对异常值不敏感)。虽然这种统计方法能够降低低秩基因的假阳性,但是这牺牲了灵敏度(功效,power)。下面我们来考虑一个低稳健性(less robust),但是通常情况下更加强大的方法。
如果我们对基个基因集中向左分布(通常来说是表达量少的基因)的基因或者说是向右分布(通常来说是就是表达高的基因)的基因戌兴趣,那么我们可以使用的最简单的统计就是比较两组之间的均值,具体可以参考这篇文献《Gene set enrichment analysis made simple》,其中G是基因在基因集中的索引,我们定义以下的统计量:
零假设是基因集中的基因表达没有差异,这些t检验的均值为0。如果t检验统计量之间互相是独立的(后面我们会学习避免此假设的方法),那么$\sqrt{N} \bar{t}$ 服从以下分布:
以下是这些统计量的qq图:
|
|
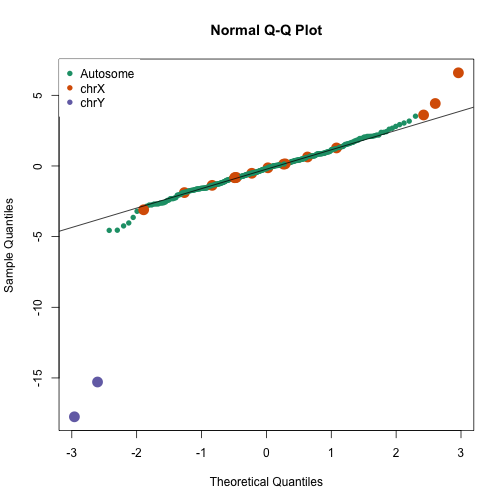
|
|
有一些算法就是基于以上原理来转统计量的均值的,例如 Tian et al 2005 , SAFE, ROAST, CAMERA.
我们可以构建其它的得分来检验其它替代假设。例如我们可以检验changes in variances 与这个分布的常规变化 [1], [2]。
基因集的假设检验
上面我们对$\sqrt{N} \bar{t}$ 的正态近似表明,t检验的统计量是独立的,并且分布相同,例如,现在我们看第二个公式,如下所示:
这个公众就是假设t检验统计量是独立的。但是,即使在零假设下,我们也许会考虑到基因集之间是相互关联的。为了了解这一点,我们可以取每个基因集中所有成对相关的平均值,我们就会看到,它们并不都是0,如下所示:
|
|
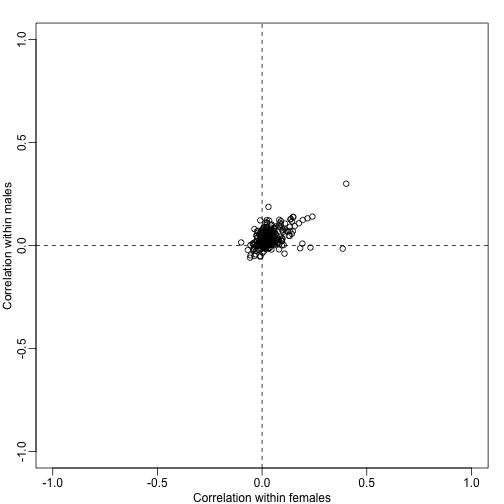
注意,在这个案例中,在一个基因集中,基因的检验统计量具有相关性的 $\rho$, 平均t检验统计量的方差如下所示:
在上面公式里,$\rho$ 是正值,方差与 $\{1 + (N-1) \rho \} $有关。虽然常规情况下,在一个基因集内,$\rho$ 并不一定都相等,但是我们仍然可以根据一个基因集内的平均 $\rho$ 来计算一个校正因子(correction factor)。这个公式可以参考这篇文献《A statistical framework for testing functional categories in microarray data》。公式中使用的校正方法还包括Wald统计量。
这些校正因子可以对上述提到的统计量进行简单的汇总。事实上,现在已经有论文提出了基于简单的均值转换方法的算法,这些算法还考虑到了相关性,它们基于渐进近似的ROAST和CAMERA,用于开发统计摘要(summary statistics)与统计推断。在计算机演示案例中,我们将会演示其中一款软件。简单起见,我们这里基于 $\sqrt{N} \bar{t}$ 计算近似,并使用校正因子。这里我们比较一下初始 $\sqrt{N} \bar{t}$ 以及校正后的 $\sqrt{N} \bar{t}$ ,如下所示:
在下图里,我们注意到这个校正会降低分数(socres)(也就是将分数值降低到接近0的程度)。尤其注意的是,第三个最高的值现在非常接近0,它不再显著(参见箭头),如下所示:
|
|
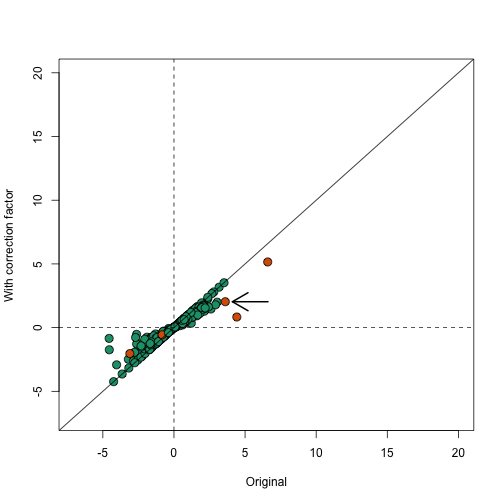
需要注意,标注出来的基因集具有相对高的相关性和大小(size),它就使得校正因子变得很大,如下所示:
|
|
在这一节中,我们已经使用了平均t检验统计量作为一个简单的案例来说明基因集在零假设下的统计摘要。我们证明了,当基因集中的基因并不独立时,需要一个校正因子。这里要注意的是,我们使用平均t检验统计量只是做了一个简单的说明,如果需要更加严格处理方法来实现均值偏移方法(testing-for-mean-shifts),则需要参考 SAFE, ROAST 和 CAMERA。
即使在使用了膨胀因子(inflation factor)校正了平均t检验统计量后,零分布也有可能不成立。例如,对于平均t检验统计量是正态的假设而言,原始的样本大小和基因集大小有可能太小。在一次部分里,我们会演示如何在不使用参数假设的情况下,使用置换检验(Permutations)来估计零分布。
置换检验(Permutations)
当我们感兴趣的结果允许以及样本足够的情况下,我们可以使用基因集特定的转换检验来替换参数检验。参考文献中列出了几种这样的方法[1], [2]。对于大多数的富集分布而言,我们可以使用置换来创建一个零分布。例如,在这个案例中,我们对简单的t检验摘要执行置换检验,如下所示:
|
|
在这个案例中,使用置换得到的结果非常类似于使用了校正因子得到的参数检验结果,我们从比较的结果中就能看出这些信息,如下所示:
|
|
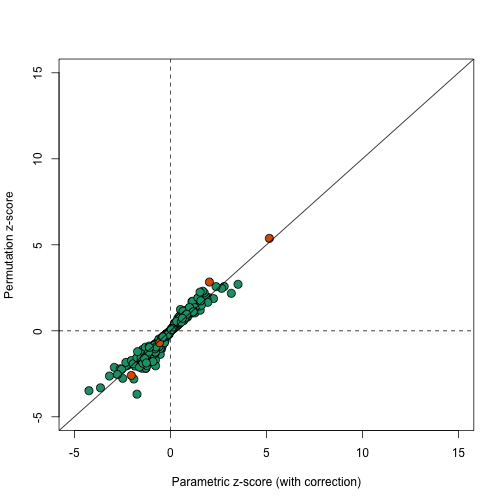
我们看到这两种方法得到的q值也是比较类似:
|
|
不过置换检验方法的一个局限就是,为了能够得到非常小的p值,我们需要进行多次置换。例如为了得到一个在10e-5到10e-6之间的一个p值,我们霜肆进行100万次置换。对于那些使用了保守的多重比较校正方法(例如Bonferonni校正)的人来说,很有必要进行更多次地的模拟。置换检验的另外一个局限就是,在更复杂的设计中,例如具有许多轭和连续协变量的实验中,如何对置换进行解读并不容易。
参考资料
- Gene set analysis
- Virtaneva K, “Expression profiling reveals fundamental biological differences in acute myeloid leukemia with isolated trisomy 8 and normal cytogenetics.” PNAS 2001 http://www.ncbi.nlm.nih.gov/pubmed/?term=11158605
- Jelle J. Goeman and Peter Bühlmann, “Analyzing gene expression data in terms of gene sets: methodological issues” Bioinformatics 2006. http://bioinformatics.oxfordjournals.org/content/23/8/980